
ЫбЫїНсЙћ: 1-15 ЙВВщЕНЁАЮяРэЛЏбЇ ЕчзгНсЙЙЁБЯрЙиМЧТМ127Ьѕ . ВщбЏЪБМф(1.912 Уы)

ЮїАВНЛЭЈДѓбЇПЦбаШЫдБбаЗЂЛљгкЕчЛЏбЇЕїПиIrO2БэУцЕчзгНсЙЙгУгкИпадФмЕчНтЫЎжЦЧтДпЛЏМСЃЈЭМЃЉ
ЮїАВНЛЭЈДѓбЇ ЕчЛЏбЇ IrO2БэУцЕчзгНсЙЙ ИпадФм ЕчНтЫЎ жЦЧт ДпЛЏМС
2020/7/1
ЕчНтЫЎБЛШЯЮЊЪЧвЛжжПЩГжајЕФЁЂМЋОпЗЂеЙЧАОАЕФВњH2ВпТдЁЃЭЈГЃЃЌЕчНтЫЎЭЈЙ§СНИіАыЗДгІНјааЃКЮібѕЗДгІЃЈOERЃЉКЭЮіЧтЗДгІЃЈHERЃЉЁЃЪЕМЪгІгУжаHERКЭOERДпЛЏМСЕФгааЇёюКЯЪЧЗЧГЃБивЊЕФЁЃОЁЙмвбБЈЕРСЫаэЖрёюКЯДпЛЏМСЃЌЕЋШдгавЛаЉЙиМќЮЪЬташвЊНјвЛВННтОіЃК1ЃЉбєМЋКЭвѕМЋЕФДпЛЏМСЭЈГЃдкГЩЗжЩЯДцдкКмДѓВювьЃЌЪЙжЦБИЙ§ГЬИДдгЃЌЧвгЩгкдкЕчНтЫЎЙ§ГЬжаПЩФмЗЂЩњДпЛЏМСШмНтЁЂдйГСЛ§ЗДгІЃЌЛсв§Ц№вѕМЋКЭбєМЋДпЛЏМСЯрЛЅИЩШХЃЛ2ЃЉДѓВПЗж...
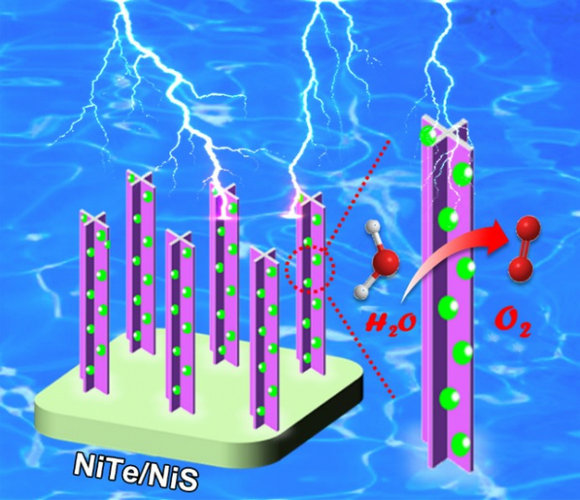
жаЩНДѓбЇЛЏбЇбЇдКРюЙтЧйНЬЪкбаОПЭХЖгЙЙНЈNiTeФЩУзеѓСаЕФНчУцЕчзгНсЙЙЕїПижњСІЕчДпЛЏбѕЮіГіЃЈЭМЃЉ
жаЩНДѓбЇЛЏбЇбЇдК РюЙтЧй НЬЪк NiTeФЩУзеѓСа НчУцЕчзгНсЙЙ ЕчДпЛЏ бѕЮіГі
2019/5/22
НќШеЃЌЮваЃЛЏбЇбЇдКРюЙтЧйбаОПЭХЖгЭЈЙ§ЙЙНЈгааЇЕФФЩУзНчУцРДЕїПиДпЛЏМСЕФЕчзгНсЙЙЃЌЩшМЦжЦБИГіИпаЇСЎМлЕФбѕЮіГіЗДгІвьжЪНсЕчДпЛЏМСЃЌВЂНсКЯЪЕбщНсЙћКЭРэТлМЦЫуЖдНчУцЕїПиДпЛЏаджЪЕФЛњжЦНјааСЫбаОПЁЃЫћУЧЪзЯШвдХнФФјЮЊЛљЕзЃЌЭЈЙ§жБНгЛЏбЇПЬЪДЕФЗНЗЈжЦБИГіХнФФјИКдиЕФЁАЪЎзжжљЁБаЭЕФNiTeФЩУзеѓСаЁЃНјвЛВНЭЈЙ§РызгНЛЛЛЗДгІдкNiTeБэУцаоЪЮNiSФЩУзПХСЃРДЕїПиNiTeЕФЕчзгНсЙЙ, жЦБИГіNiTe/NiS вьжЪНсДпЛЏМСЁЃ...
ЮЊСЫИќПЩППЕиЕУЕНЫЎШмвКжаЕААзжЪЗжзгЕФЕчзгНсЙЙ,гаБивЊЙЙНЈЫЎШмвКЖдЕААзжЪЗжзгЕчзгНсЙЙЕФЕШаЇЪЦ,етИіЕШаЇЪЦБиаыМђЕЅЁЂвзгУ.ЭЈЙ§ЕквЛаддРэЁЂШЋЕчзгЁЂДгЭЗМЦЫу,ЙЙдьСЫЫЎШмвКЖдРвАБЫс(Tyr)ЕчзгНсЙЙЕФЕШаЇЪЦ.ЙЄзїЗжЮЊШ§ВН:(1)гУЁАздгЩЭХДиМЦЫуЗЈЁБМЦЫувЛИіКЌРвАБЫсКЭЫЎЗжзгЕФЯЕЭГЕФФмСПзюЕЭЪБЕФПеМфНсЙЙ;(2)ЛљгкЕквЛВНЕФПеМфНсЙЙ,гУЁАЭХДиТёШыздЧЂМЦЫуЗЈЁБМЦЫуРвАБЫсдквдЫЎЗжзгЮЊЭтЪЦЬѕМўЯТЕФЕчзгНсЙЙ;(3)гУЁА...
14зхдгЛЗЮьЖўЯЉЗжзг(ЙшЁЂерЁЂЮ§)ЕФЕчзгНсЙЙгыЙтЦзаджЪ
УмЖШЗККЏРэТл ЙшдгЛЗЮьЖўЯЉ еёЖЏЙиСЊКЏЪ§ ЙтЮќЪе ЙтЗЂЩф
2010/3/9
14зхдгдзгШЁДњЕФдгЛЗЮьЖўЯЉЗжзгОпгаЖРЬиЕФЙтЦзаджЪ, ГЩЮЊЗЂЙтВФСЯЕФУїаЧЗжзг. ЮЊСЫИќЩюВуДЮЕиРэНтЙшЁЂерЁЂЮ§дгЛЗЮьЖўЯЉЗжзгЕФЙтЦзаджЪ, БОЮФДгРэТлЩЯМЦЫуСЫЫќУЧЕФЕчзгНсЙЙМАЦфЮќЪеКЭЗЂЩфЙтЦз. ЗжБ№ВЩгУУмЖШЗККЏРэТл(DFT)КЭКЌЪБУмЖШЗККЏРэТл(TD-DFT), гХЛЏСЫЙшЁЂерЁЂЮ§дгЛЗЮьЖўЯЉЗжзгЛљЬЌКЭЕквЛМЄЗЂЬЌЕФЦНКтЙЙаЭ, МЦЫуСЫЕчзгНсЙЙКЭеёЖЏаджЪ. дкДЫЛљДЁЩЯ, дЫгУеёЖЏЙиСЊКЏЪ§ЙЋЪНМЦЫуСЫЮќЪеЙтЦзКЭЗЂЩф...
ВЩгУЕквЛаддРэЦНУцВЈиЭЪЦМЦЫуСЫКЌгаПеЮЛШБЯнЕФЛЦЬњПѓЕФЕчзгНсЙЙ, ЭЌЪБЬжТлСЫПеЮЛШБЯнЖдЛЦЬњПѓИЁбЁааЮЊЕФгАЯь. баОПНсЙћБэУї, СђПеЮЛЖдОЇАћЬхЛ§гАЯьВЛДѓ, ЬњПеЮЛЪЙЛЦЬњПѓОЇАћХђеЭСЫ1.29%. ПеЮЛШБЯнжївЊгАЯьЛЦЬњПѓЗбУзФмМЖИННќЕФЕчзгФмДјНсЙЙ, ВЂдкНћДјжаГіЯжСЫаТФмМЖ. ПеЮЛЕФДцдкЪЙЛЦЬњПѓЕФЗбУзФмМЖЩ§Ип, ВЛРћгкЛЦЬњПѓЕФИЁбЁ. СэЭт, гааЇжЪСПМЦЫуБэУї, ПеЮЛЕФДцдкдіЧПСЫЛЦЬњПѓЕМДјЕзЕчзгЕФЖЈгђад. M...
ЮЊбаОПSbВєдгЖдTi/SnO2ЕчМЋЮШЖЈадгыЕМЕчадЕФгАЯь, ВЩгУЛљгкУмЖШЗККЏРэТлЕФЦНУцВЈиЭЪЦЗНЗЈЖдН№КьЪЏаЭSnO2МАВЛЭЌБШР§SbВєдгSnO2ЬхЯЕНјааСЫЕквЛаддРэМЦЫу, гУЙувхЬнЖШНќЫЦЗНЗЈгХЛЏСЫSn1-xSbxO2ЙЬШмЬхЕчМЋЕФОЇЬхНсЙЙ, МЦЫуСЫВєдгЧАКѓЬхЯЕЕФЕчзгНсЙЙвдМАВЛЭЌВєдгБШР§ЪБЕФаЮГЩФм. НсЙћБэУї: SbЬцДњSnКѓ, ОЇИёГЃЪ§гыОЇАћЬхЛ§ОљдіМг, ЕЋВєдгаЮГЩФмЫцВєдгСПБфЛЏВЛДѓ, дкВєдгСПЮЊ0.08...
ВЩгУУмЖШЗККЏРэТлМАиЭЪЦЦНУцВЈЗНЗЈ, ЖдЮДВєдгSnO2вдМАЙ§ЖЩН№ЪєVЁЂCrЁЂMnВєдгSnO2ЕФГЌдАћЬхЯЕНјааСЫМИКЮгХЛЏ, МЦЫуСЫОЇИёГЃЪ§ЁЂЕчзгНсЙЙгыДХбЇаджЪ. НсЙћБэУї, 6.25%гы12.5%СНжжВєдгХЈЖШЪБ, ЬхЯЕЕФЕчзгзда§КЭДХбЇаджЪУЛгаЗЂЩњКмДѓЕФБфЛЏ; ЯрЖдгкЮДВєдгSnO2, Й§ЖЩН№ЪєВєдгКѓSnO2жаOдзггаЯђЙ§ЖЩН№ЪєвЦЖЏЕФЧїЪЦ, ВЂЪЙЕУOгыВєдгН№ЪєжЎМфМќГЄБфЖЬ; дкVКЭCrВєдгКѓ, SnO2Оп...
[M(N)X2]-(M=Ru, Os; X=S2C6H4, mnt)ЕФЕчзгНсЙЙКЭЙтЦзаджЪЕФРэТлбаОП
ЕЊЛЏюЩХфКЯЮя ЕЊЛЏяАХфКЯЮя ЮќЪеЙтЦз УмЖШЗККЏРэТл КЌЪБУмЖШЗККЏРэТл
2010/1/19
РћгУУмЖШЗККЏРэТл(DFT)жаЕФB3LYPЗНЗЈгХЛЏСЫЕЊЛЏюЩКЭЕЊЛЏяАХфКЯЮя[M(N)X2]-[M=Ru, Os; X=S2C6H4, mnt(maleonitriledithiolate)]ЕФЛљЬЌМИКЮНсЙЙ, ЕУЕНЕФМИКЮВЮЪ§гыЪЕбщНсЙћЮЧКЯЕУКмКУ. ВЩгУTD-DFTЗНЗЈ, ЕУЕНСЫХфКЯЮядкCH3CNШмвКжаЕФМЄЗЂЬЌЕчзгНсЙЙКЭЕчзгЮќЪеЙтЦз. РћгУSCRFЗНЗЈжаЕФCPCMФЃаЭРДФЃФтШмМСЛЏаЇгІ. баОПНсЙћБэУї, Хф...
4-(1,2-ЖўБНЛљ)ввЯЉЛљ-4ЁЏ-(N,N-ЖўБНЛљ-4-ввЯЉЛљБНАЗЛљ)СЊБНМАЦфЖўЗњШЁДњбмЩњЮяЕФЕчзгНсЙЙгыЙтЦзаджЪ
СЊБНввЯЉЛљбмЩњЮя УмЖШЗККЏРэТл ПебЈзшЕВ ЙтЦзаджЪ ЕчзгНсЙЙ
2010/1/14
ЗжБ№ВЩгУB3LYP/6-31G(d)КЭCIS/6-31G(d)ЗНЗЈЖд4-(1,2-ЖўБНЛљ)ввЯЉЛљ-4ЁЏ-(N,N-ЖўБНЛљ-4-ввЯЉЛљБНАЗЛљ)СЊБН(A)МАЦфЖўЗњШЁДњбмЩњЮя(B-F)ЕФЛљЬЌ(S0)КЭЕЅжиМЄЗЂЬЌ(S1)ЕФМИКЮЙЙаЭНјааСЫШЋгХЛЏ, МЦЫуЛёЕУСЫЕчРыЪЦIPЁЂЕчзгЧзКЭЪЦEAЕШЯрЙиЪ§Он, ВЂВЩгУКЌЪБУмЖШЗККЏ(TD-DFT)ЗНЗЈМЦЫуСЫЩЯЪіЛЏКЯЮяЕФЕчзгЮќЪеКЭгЋЙтЗЂЩфЙтЦз. баОПНсЙћБэУї, ЛЏКЯЮяAМАЖўЗњ...
ЛљгкЙувхЬнЖШНќЫЦ(GGA)ЕФУмЖШЗККЏРэТл(DFT), ЭЈЙ§ЙЙдьЬњДХ(FM), зшДьЕФШ§НЧЗЧЙВЯпЗДЬњДХ(FAFM)ЁЂЩЯЩЯЯТЯТаЭЙВЯпЗДЬњДХ(ЁќЁќЁ§Ё§AFM)Ш§жжВЛЭЌДХадЙЙаЭ, ДгЗЧЙВЯпДХадНсЙЙМЦЫуГіЗЂ, гХЛЏСЫЕЭЮТЭЬњПѓCuFeO2ОЇЬхВФСЯЕФМИКЮНсЙЙ, баОПСЫДХадНсЙЙЖдЕчзгНсЙЙЁЂФмЯЖКЭДХОиЕШЕФзїгУ. МЦЫуЗЂЯжЩЯЩЯЯТЯТаЭЗДЬњДХзда§ХХСаФмДйНјФмЯЖаЮГЩ, змФмНЕЕЭ, ДХОидіДѓ. гЩгкЩЯЩЯЯТЯТаЭЗДЬњДХгызшДьШ§...
Pu3MКЭPuM3 (M=Ga, In, Sn, Ge)ЛЏКЯЮяЕФЕчзгНсЙЙКЭаЮГЩШШ
ШЋЪЦЯпадзКМгЦНУцВЈ Pu3MКЭPuM3ЛЏКЯЮя ЕчзгНсЙЙ аЮГЩШШ
2010/1/6
ВЩгУШЋЪЦЯпадзКМгЦНУцВЈ(FPLAPW)ЗНЗЈ, дкЙувхЬнЖШНќЫЦ(GGA)+зда§ЙьЕРёюКЯ(SOC)+зда§МЋЛЏ(SP)ЯТМЦЫуСЫОпгаAuCu3ЙЙаЭЕФPu3MКЭPuM3 (M=Ga, In, Sn, Ge)ЛЏКЯЮяЕФЦНКтНсЙЙЁЂЕчзгНсЙЙКЭаЮГЩШШ. МЦЫуЕФОЇИёГЃЪ§гыЪЕбщжЕЗћКЯЕУКмКУ; ЬЌУмЖШЗжЮіБэУїPu КЭM дзгЙьЕРМфЕФдгЛЏзїгУОіЖЈгкPu 6d-Pu 5fЁЂMp-Pu 6dКЭMsp-M spЙьЕРдгЛЏжЎМфЕФОКељ,...
ВЩгУУмЖШЗККЏРэТл(DFT)дкЙувхЬнЖШНќЫЦ(GGA)ЯТЕФЦНУцВЈГЌШэиЭЪЦЗЈ, баОПСЫSr2-xLaxCrReO6(x=0, 0.25, 0.5, 1)ЕФОЇЬхНсЙЙЁЂЕчзгНсЙЙКЭДХад. ЭЈЙ§МИКЮНсЙЙгХЛЏ, ЕУЕНСЫВФСЯЕФОЇИёГЃЪ§ЁЂЕчзгКЭзда§ЗжВМвдМАДХОиЕФДѓаЁ. ЗжЮіСЫLaЕчзгВєдгЖдSr2CrReO6ВФСЯНсЙЙЕФгАЯь, ЗЂЯжЕБLaВєдгХЈЖШНЯаЁ(x<1)ЪБ, Sr2-xLaxCrReO6ШдБЃГжАыН№ЪєЬиад, ЕЋИеКУдк...
ВЩгУЛљгкУмЖШЗККЏРэТлЕФЕквЛаддРэЦНУцВЈГЌШэиЭЪЦМЦЫуЗНЗЈ, баОПСЫInЁЂSc pаЭВєдгЖдSrTiO3ФИЬхЛЏКЯЮяЮШЖЈадЁЂЕчзгНсЙЙКЭЙтбЇаджЪЕФгАЯь. МЦЫуНсЙћБэУї:ВєдгКѓ, SrIn0.125Ti0.875O3КЭSrSc0.125Ti0.875O3ЕФЮШЖЈадНЕЕЭ, ЬхЯЕЯдЪОpаЭМђВЂАыЕМЬхЬиеї, ВєдгНів§Ц№дгжЪдзгНќСкЧјгђЕФМИКЮНсЙЙЗЂЩњБфЛЏ. ЭЌЪБ, SrIn0.125Ti0.875O3КЭSrSc0.125...
дкУмЖШЗККЏРэТл(DFT)ЕФB3LYPЫЎЦНЖдВЛЭЌЭщЛљдкВЛЭЌЮЛжУШЁДњаЮГЩЕФ8жжТоЕЄУїЛЏКЯЮяНјааНсЙЙгХЛЏ, ВЂдкДЫЛљДЁЩЯгІгУКЌЪБУмЖШЗККЏРэТл(TD-DFT)КЭЕЅМЄЗЂзщЬЌЯрЛЅзїгУ(CIS)ЗНЗЈЗжЮіСЫШЁДњЛљЖдТоЕЄУїЕФЕчзгНсЙЙЁЂЧАЯпЗжзгЙьЕРМАЕчзгЙтЦзЕФгАЯь. МЦЫуНсЙћБэУї, ЧАЯпЗжзгЙьЕРжївЊЗжВМдкТоЕЄУїЗжзгЕФбѕдгньЛЗЩЯ, ТоЕЄУїЗжзгжаСНИіNЖЫЕФHИїжЛга1ИіHБЛЭщЛљШЁДњЪБ, зюИпеМОнЙьЕР(HOMO)дкжївЊЙВщюЛЗ...
РћгУЕчКЩздЧЂРыЩЂБфЗжXІС(SCC-DV-XІС)ЗНЗЈМЦЫуСЫЗАЛљЙЬШмЬхжаЗАЧтЗДгІЧАКѓЗАМАЦфЧтЛЏЮя(VHx, x=0, 1, 2)ЁЂМйЖЈдзгДиФЃаЭVHx*(x=1, 2)КЭVHxЁф(x=0, 1)ЕФЕчзгНсЙЙ. НсЙћБэУїЃКЗАгыЧтЦјЗДгІЩњГЩVHЪБ, ЛЏбЇаЇгІКЭНсЙЙаЇгІЖМЪЙV 3dКЭH 1sЙьЕРЯђЕЭФмСПЗНЯђвЦЖЏ, ЧтЛЏЮяVHжаV 3dКЭH 1sЙьЕРжиЕўзюЖр, VЃHжЎМфЕФЯрЛЅзїгУНЯЧПЁЃVHдйгыЧтЦјЗДгІЩњГЩVH...
жаЙњбаОПЩњНЬг§ХХааАё-Ьѕ
- е§дкМгди...
жаЙњбЇЪѕЦкПЏХХааАё-Ьѕ
- е§дкМгди...
ЪРНчДѓбЇПЦбаЛњЙЙХХааАё-Ьѕ
- е§дкМгди...
жаЙњДѓбЇХХааАё-Ьѕ
- е§дкМгди...
ШЫЁЁЮя-ЦЊ
- е§дкМгди...
ПЮЁЁМў-ЦЊ
- е§дкМгди...
ЪгЬ§зЪСЯ-ЦЊ
- е§дкМгди...
баеазЪСЯ -ЦЊ
- е§дкМгди...
жЊЪЖвЊЮХ-ЦЊ
- е§дкМгди...
ЙњМЪЖЏЬЌ-ЦЊ
- е§дкМгди...
ЛсвщжааФ-ЦЊ
- е§дкМгди...
бЇЪѕжИФЯ-ЦЊ
- е§дкМгди...
бЇЪѕеОЕу-ЦЊ
- е§дкМгди...