
�������: 106-120 ���鵽��ԭ������Ӷ���ѧ����ؼ�¼161�� . ��ѯʱ��(0.744 ��)
��˫�����Ŵ�����������Ԫ�ر����ܵIJ�����Ŵ��γ�ʱ����ƫ��, �Ӷ�Ӱ���Ŵص��ۻ���ԭ���Ų��ͽṹ. ���˫�����Ŵ���ԭ��ƫ����Ϊ���о�����Ϊ�������ṹ�Ŀɿ��Ʊ��ṩ���ۻ���. �����÷��Ӷ���ѧ���Ƕ��ԭ�ӷ����о��˷ֲ����Ŵصĺ��IJ���DZ�����Ag ԭ��ƫ����(AgPd)147 �Ŵ��ۻ���Ӱ��. �������Ag ��Pd �ŴصIJ�ͬλ��ʱ������ͬ,�ڱ����ʱ���ȶ�, ����Ǻ��IJ�, ������DZ���...
���÷�ƽ����Ӷ���ѧ������S i/Ge������ṹ�Ľ����������ģ�⡣ģ�����������������ԡ�Ľ���������������ṹ���ȴ�������,���ҳ�����ṹ�����ڳ����Լ��¶ȵ�����Ҳ��Խ������������ͬӰ�졣�������ڳ��ȵ�����,��������Խ��ԽС,������ṹ�ĵ���������ǿ�������ӷǵ���ɢ����Ƶ�Ӱ��,�����������¶ȵ�����Ҳ��С��
�й���ѧԺ���������о�������ԭ����ײ��Ӧ����ѧ�о�ȡ����Ҫ��չ��ͼ��
�й���ѧԺ���������о��� ����ԭ����ײ ��Ӧ����ѧ
2011/10/19
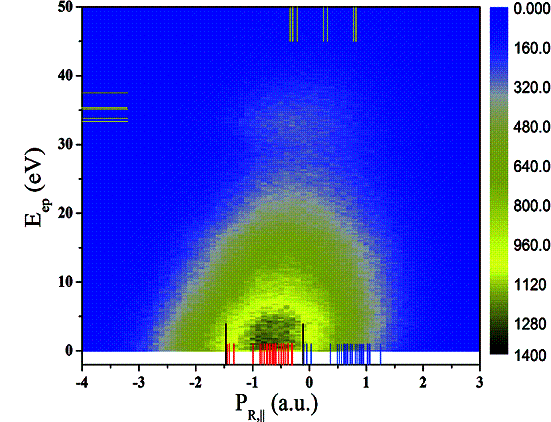
�й���ѧԺ���������о���ԭ������һ�������Ա�����������Ƶķ�Ӧ���������ǣ��о���30keV/u��He2+ ������Arԭ����ײʵ���еĵ�����ͬʱ�����뷴Ӧͨ���������Ҫ��չ��
������һ������װ�ò�������̬��O2(1��g)��O2���������Ľ���Cu��Cr��Ni��Ag����������. ��ʵ��ʱ���O2(1��g)Ũ�����ӣ����������. ������������Ʒ�ڼ���˹������ն��±�¶��Сʱ������ʻ�ص�ԭ��ˮƽ. �����һ�ֻ��ڱ�������λ������ѧ�����ı�������������O2(1��g
����Phyre���������, ����������øEstTs1����ά�ṹ, ��ͨ�����Ӷ���ѧ�Ż�����, �õ��˿ɿ��Ĺ���. ���ӶԽ��о�����, ��p������������������EstTs1�����ʵ���, ���С���ʺ�EstTs1�Ļ��Կڴ�. Thr111�ǵ�����ø��ϵ���Ҫ�л�, ������γ������; Ser85����Ҫ�Ĵ��л�.
�÷��Ӷ���ѧ�����о��˸�����̼���ܱ���Ľ�ԭ�����γɵĽṹ�������ȶ���. �о��������,��ԭ����̼���ܱ������γɶ������ȶ��Ľṹ������ԭ�Ӹ����ʵ���15.91% ʱ,�γɽ�������; ��ԭ�Ӹ�����Ϊ15.91%~104.98% ʱ,�γ�һϵ�еĿDz�ṹ; ����ԭ�Ӹ����ʴ���104.98% ʱ,����־��������������ṹ. �ڶ���̼����֧�ŵĽ����Dz�ṹ�����ȶ��Խ����о���, �������۵����...
���÷��Ӷ���ѧ�����о��˺��ܲ��ϻ����Ǽ�������(RDX)��3-������-3-��������(AMMO)��RDX/AMMO�ƽ���. �������AMMO��RDX(010)�����֮���������ǿ�������(100)��(001)�森����غ���g(r)������RDX��AMMO֮�������ã�������RDX/AMMO�ƽ����ĵ���ϵ��������ѹ�����ɱȵ����ܣ��о����������AMMO�ļ����ܹ�����RDX�ĵ�����ѧ���ܣ����ǵ�...
����H(D)ԭ����±�̬ʱ��ɶ������о���HOD����������������124 nm������?̬����ѧ��ʵ�������HOD������?̬��ת���ֱ�������ף����õ������ת������H+OD�Լ�D+OHͨ���IJ����ܵ�ƽ�����ף�ͨ���Բ���ƽ�����ķ������õ���?̬�������OH��OD�ķ�֧�ȣ����릢̬��?̬��Ӧ�������ķ�֧�����˶Աȣ�ʵ����ȷ����HOD����OD���Ľ�����Ϊ41751.3��5 cm-1.
�й���ѧԺ�人��������ѧ�о����ڷ��뼤�ⳡԭ�ӷ��Ӷ���ѧ�о�����ȡ����Ҫ��չ
�й���ѧԺ�人��������ѧ�о��� ���뼤�ⳡԭ�ӷ��Ӷ���ѧ ��Ҫ��չ
2010/6/13
ǿ���뼤���յ������ĵ�����������г���ʱ�䣨�Ƿ���߶ȣ��Ϳռ䣨�ǰ��߶ȣ��ֱ���������һ��̽��ԭ�ӷ����ڲ��ṹ�����춯��ѧ�ݻ����������ߡ�ԭ�ӷ�����ǿ���뼤�ⳡ�еĸ߽����ϵ��������ɵ��������ĸ�˷���������ײ�������ڵ������Я���˳�ʼԭ�ӷ��ӽṹ��Ϣ����˿������ø߽����ϵ�������ĵ����������Ƿֲ�̽��ԭ�ӷ��ӵĽṹ�����춯��ѧ�ݻ���Ȼ������ĿǰΪֹ�������ձ���Ϊ�Է��ӽṹ�ij�������о�ֻ���ڶԷ�...
��������Ӷ���ѧ���̵����Dz�ֵ��
������Ӷ���ѧ ȫ��ʽ ���Dz�ֵ ����
2011/8/17
����������Ӷ���ѧ�������У��������ܶ�N��t��ʹ�����Ǻ������Ʒ�������ʹ��ȫ��ʽ��ʽ��֤A�ȶ��ԣ�����һ�����ķ������������������˷����������Ӧ���ڸ��ַ�Ӧ�����룬����ʱ��̣����㾫�Ƚϸߣ���������ʵ�ʹ��̵���Ҫ�������㶯̬���̵�һ�ֽϺ÷�����
Һ̬�����ṹ�仯�ķ��Ӷ���ѧģ���о�
Һ̬���� �ṹת�� ���Ӷ���ѧģ��
2009/12/28
Һ̬�����ṹ�仯�ķ��Ӷ���ѧģ���о���
ԭ�ӳߴ������Ǿ��γ�����
���Ӷ���ѧģ�� �Ǿ��;��� ԭ�ӳߴ粻ƥ��
2009/12/28
���÷��Ӷ���ѧģ�⼼�����о��˴�Au��AuCu�Ͻ���ۻ����Ǿ����;�������.ģ��������,����ȴ����Ϊ5��1011 K•s��1��4��1012 K•s��1�ķ�Χ�ڣ�Һ̬Au�����γɾ��壬������Խ�죬�ᾧ�¶�Խ�ͣ���AuCu�Ͻ����γɷǾ���������Խ�죬�Ǿ�ת���¶�Խ��.��֤��ԭ�ӳߴ�IJ�ƥ�������ڷǾ��γ���һ����.
ͨ�����Ӷ���ѧģ�⣬�о���������ҺNaCaF_3��Na_2CaF_4��Na_4CaF_5��ϵ��ģ����������ֶ�Ԫ���ϵ�ľ���ֲ�����ʮ�ֽӽ�����ģ�����õ���Ħ������ʺܺõ���ʵ��ֵһ�£��������Na~+�����������֮����ֳ��ܺõ����Թ�ϵ��ģ���������Na_2CaF_4��ϵ�У���NaF-CaF_2��Ԫϵ���ڵ��ۻ����ֱ�NaF:CaF_2��2:1ʱ��Na~+��Ca~(2+)��F~-���ӵ�����ɢϵ...
RbCl�۽�ķ��Ӷ���ѧģ���о�
RbCl �۽���� ��ѹ���Ӷ���ѧģ��
2009/12/8
���õ�ѹ���Ӷ���ѧģ�ⷽ�����о��˴Ӿ��ൽҺ�ͬ�¶���RbCl��ϵ�Ľṹ�����ʣ���ѹģ�������ȿ�ģ��һ�µ�ƽ�����ʡ��������ʺͽṹ�����������������������ڲ��Ƽ����ȹ��������£���ϵ����ԥʱ������������ۻ����̣����ڲ���Ҫ�ܶ����ݣ���ѹģ��������չ��Ϊ��������е�һ���ֶ�.
���ô���ģ���˻����ַ��Ӷ���ѧ�ķ������������Է��Ľ����˹��������������˶��Gly��Phe�£���ת�ǵĵ��ܹ����������������´���ģ���˻����߽������������Ч��Ѱ�ҵ��ܹ���
�й��о����������а�-��
- ���ڼ���...
�й�ѧ���ڿ����а�-��
- ���ڼ���...
�����ѧ���л������а�-��
- ���ڼ���...
�й���ѧ���а�-��
- ���ڼ���...
�ˡ���-ƪ
- ���ڼ���...
�Ρ���-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
�������� -ƪ
- ���ڼ���...
֪ʶҪ��-ƪ
- ���ڼ���...
���ʶ�̬-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
ѧ��ָ��-ƪ
- ���ڼ���...
ѧ��վ��-ƪ
- ���ڼ���...